


Journal of Computer Applications ›› 2024, Vol. 44 ›› Issue (12): 3958-3964.DOI: 10.11772/j.issn.1001-9081.2023121846
• Frontier and comprehensive applications • Previous Articles
Changjiu HE1,2, Jinghan YANG2, Piyu ZHOU2, Xinye BIAN1, Mingming LYU1, Di DONG1, Yan FU2, Haipeng WANG1( )
)
Received:2024-01-05
Revised:2024-03-25
Accepted:2024-04-02
Online:2024-04-15
Published:2024-12-10
Contact:
Haipeng WANG
About author:HE Changjiu, born in 1997, M. S. candidate. His research interests include deep learning, bioinformatics.Supported by:
何长久1,2, 杨婧涵2, 周丕宇2, 边昕烨1, 吕明明1, 董迪1, 付岩2, 王海鹏1()
通讯作者:
王海鹏
作者简介:何长久(1997—),男,山东淄博人,硕士研究生,主要研究方向:深度学习、生物信息学基金资助:CLC Number:
Changjiu HE, Jinghan YANG, Piyu ZHOU, Xinye BIAN, Mingming LYU, Di DONG, Yan FU, Haipeng WANG. Theoretical tandem mass spectrometry prediction method for peptide sequences based on Transformer and gated recurrent unit[J]. Journal of Computer Applications, 2024, 44(12): 3958-3964.
何长久, 杨婧涵, 周丕宇, 边昕烨, 吕明明, 董迪, 付岩, 王海鹏. 基于Transformer和门控循环单元的肽序列理论串联质谱图预测方法[J]. 《计算机应用》唯一官方网站, 2024, 44(12): 3958-3964.
Add to citation manager EndNote|Ris|BibTeX
URL: https://www.joca.cn/EN/10.11772/j.issn.1001-9081.2023121846
| 数据集编号 | 物种 | 实验室 | 使用的能量值 | 谱图数 |
|---|---|---|---|---|
| PXD004732[ | 合成 | Kuster | 20,23,25,28,30,35 | 831 328 |
| PXD001468[ | 人 | Gygi | 25 | 35 404 |
| PXD000269[ | 酵母 | Mann | 25 | 66 008 |
| PXD001250[ | 鼠 | Mann | 25,27 | 102 719 |
| PXD004584[ | 线虫 | Kenyon | 25 | 50 911 |
Tab. 1 Information of datasets
| 数据集编号 | 物种 | 实验室 | 使用的能量值 | 谱图数 |
|---|---|---|---|---|
| PXD004732[ | 合成 | Kuster | 20,23,25,28,30,35 | 831 328 |
| PXD001468[ | 人 | Gygi | 25 | 35 404 |
| PXD000269[ | 酵母 | Mann | 25 | 66 008 |
| PXD001250[ | 鼠 | Mann | 25,27 | 102 719 |
| PXD004584[ | 线虫 | Kenyon | 25 | 50 911 |

Fig. 1 Heatmap of MS2 similarity at different NCEs

Fig. 2 Model design
| 离子类型 | PCC>0.70 | PCC>0.75 | PCC>0.80 | PCC>0.85 | PCC>0.90 |
|---|---|---|---|---|---|
| 18种离子 | 99.49 | 98.99 | 98.15 | 96.22 | 92.15 |
| b系列 | 96.46 | 94.87 | 93.13 | 90.22 | 84.42 |
| y系列 | 99.40 | 99.15 | 98.60 | 97.64 | 95.16 |
| a系列 | 88.49 | 86.11 | 83.30 | 79.24 | 72.73 |
Tab. 2 Percentage of PCC metric of different ions
| 离子类型 | PCC>0.70 | PCC>0.75 | PCC>0.80 | PCC>0.85 | PCC>0.90 |
|---|---|---|---|---|---|
| 18种离子 | 99.49 | 98.99 | 98.15 | 96.22 | 92.15 |
| b系列 | 96.46 | 94.87 | 93.13 | 90.22 | 84.42 |
| y系列 | 99.40 | 99.15 | 98.60 | 97.64 | 95.16 |
| a系列 | 88.49 | 86.11 | 83.30 | 79.24 | 72.73 |
| 离子类型 | 肽序列长度 | ||||
|---|---|---|---|---|---|
| ≤10 | 11~15 | 16~20 | 21~25 | ≥26 | |
| 18种离子 | 0.990 | 0.982 | 0.968 | 0.951 | 0.931 |
| b系列 | 0.992 | 0.982 | 0.961 | 0.942 | 0.912 |
| y系列 | 0.993 | 0.988 | 0.979 | 0.972 | 0.953 |
| a系列 | 0.996 | 0.978 | 0.929 | 0.875 | 0.815 |
Tab. 3 PCC mid-value distribution in peptide sequences of different lengths
| 离子类型 | 肽序列长度 | ||||
|---|---|---|---|---|---|
| ≤10 | 11~15 | 16~20 | 21~25 | ≥26 | |
| 18种离子 | 0.990 | 0.982 | 0.968 | 0.951 | 0.931 |
| b系列 | 0.992 | 0.982 | 0.961 | 0.942 | 0.912 |
| y系列 | 0.993 | 0.988 | 0.979 | 0.972 | 0.953 |
| a系列 | 0.996 | 0.978 | 0.929 | 0.875 | 0.815 |

Fig. 3 Comparison of predicted and experimental spectrums of EVLEGLEYLHK(2+)
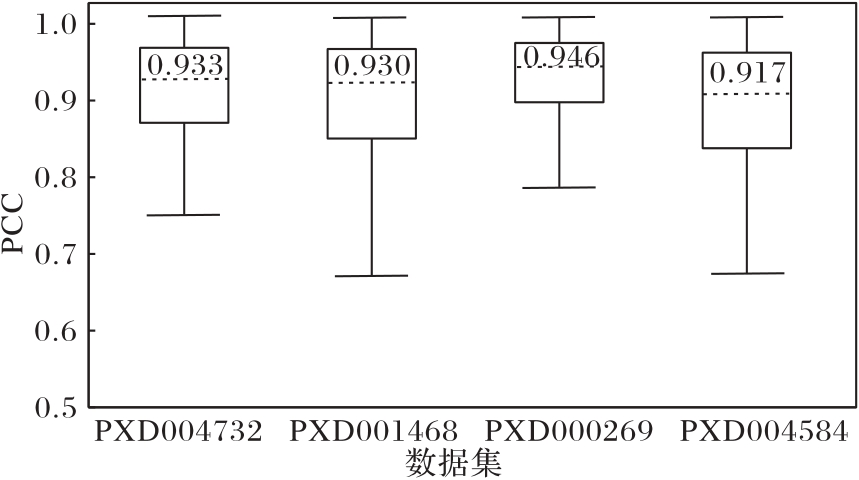
Fig. 4 PCC distribution of cross-laboratory and cross-species tests

Fig. 5 PCC distribution of cross-NCE tests
| 模型 | PCC>0.70 | PCC>0.75 | PCC>0.80 | PCC>0.85 | PCC>0.90 |
|---|---|---|---|---|---|
| pDeep默认模型 | 93.84 | 92.87 | 91.47 | 89.59 | 86.20 |
| pDeep_re模型 | 96.68 | 96.31 | 95.26 | 93.58 | 90.03 |
| DeepCollider模型 | 99.08 | 98.71 | 98.00 | 96.66 | 93.19 |
Tab. 4 Metric comparison of three models
| 模型 | PCC>0.70 | PCC>0.75 | PCC>0.80 | PCC>0.85 | PCC>0.90 |
|---|---|---|---|---|---|
| pDeep默认模型 | 93.84 | 92.87 | 91.47 | 89.59 | 86.20 |
| pDeep_re模型 | 96.68 | 96.31 | 95.26 | 93.58 | 90.03 |
| DeepCollider模型 | 99.08 | 98.71 | 98.00 | 96.66 | 93.19 |
| 指标 | 方法 | PXD001468 | PXD000269 | PXD001250 | PXD004584 |
|---|---|---|---|---|---|
PCC 均值 | pDeep | 0.668 | 0.812 | 0.781 | 0.818 |
| Prosit | 0.662 | 0.812 | 0.775 | 0.813 | |
| DeepCollider | 0.847 | 0.918 | 0.883 | 0.890 | |
PCC 中值 | pDeep | 0.615 | 0.770 | 0.738 | 0.752 |
| Prosit | 0.612 | 0.770 | 0.732 | 0.747 | |
| DeepCollider | 0.774 | 0.888 | 0.857 | 0.838 | |
MAE 均值 | pDeep | 0.022 | 0.020 | 0.019 | 0.016 |
| Prosit | 0.022 | 0.020 | 0.020 | 0.017 | |
| DeepCollider | 0.017 | 0.015 | 0.014 | 0.013 | |
MAE 中值 | pDeep | 0.023 | 0.020 | 0.021 | 0.018 |
| Prosit | 0.023 | 0.020 | 0.022 | 0.019 | |
| DeepCollider | 0.019 | 0.015 | 0.016 | 0.015 |
Tab.5 Comparison of PCC and MAE on different datasets
| 指标 | 方法 | PXD001468 | PXD000269 | PXD001250 | PXD004584 |
|---|---|---|---|---|---|
PCC 均值 | pDeep | 0.668 | 0.812 | 0.781 | 0.818 |
| Prosit | 0.662 | 0.812 | 0.775 | 0.813 | |
| DeepCollider | 0.847 | 0.918 | 0.883 | 0.890 | |
PCC 中值 | pDeep | 0.615 | 0.770 | 0.738 | 0.752 |
| Prosit | 0.612 | 0.770 | 0.732 | 0.747 | |
| DeepCollider | 0.774 | 0.888 | 0.857 | 0.838 | |
MAE 均值 | pDeep | 0.022 | 0.020 | 0.019 | 0.016 |
| Prosit | 0.022 | 0.020 | 0.020 | 0.017 | |
| DeepCollider | 0.017 | 0.015 | 0.014 | 0.013 | |
MAE 中值 | pDeep | 0.023 | 0.020 | 0.021 | 0.018 |
| Prosit | 0.023 | 0.020 | 0.022 | 0.019 | |
| DeepCollider | 0.019 | 0.015 | 0.016 | 0.015 |
| 方法 | PXD001468 | PXD000269 | PXD001250 | PXD004584 | ||||
|---|---|---|---|---|---|---|---|---|
| PCC>0.70 | PCC>0.90 | PCC>0.70 | PCC>0.90 | PCC>0.70 | PCC>0.90 | PCC>0.70 | PCC>0.90 | |
| pDeep | 44.73 | 11.28 | 72.35 | 23.79 | 65.04 | 19.44 | 67.71 | 29.11 |
| Prosit | 44.89 | 10.25 | 73.57 | 21.58 | 66.94 | 16.04 | 67.47 | 26.53 |
| DeepCollider | 70.88 | 36.67 | 95.45 | 59.22 | 91.81 | 42.83 | 84.18 | 47.13 |
Tab.6 Proportions of PCC>0.70 and PCC>0.90 on different datasets
| 方法 | PXD001468 | PXD000269 | PXD001250 | PXD004584 | ||||
|---|---|---|---|---|---|---|---|---|
| PCC>0.70 | PCC>0.90 | PCC>0.70 | PCC>0.90 | PCC>0.70 | PCC>0.90 | PCC>0.70 | PCC>0.90 | |
| pDeep | 44.73 | 11.28 | 72.35 | 23.79 | 65.04 | 19.44 | 67.71 | 29.11 |
| Prosit | 44.89 | 10.25 | 73.57 | 21.58 | 66.94 | 16.04 | 67.47 | 26.53 |
| DeepCollider | 70.88 | 36.67 | 95.45 | 59.22 | 91.81 | 42.83 | 84.18 | 47.13 |
| 1 | 孙瑞祥,付岩,李德泉,等. 基于质谱技术的计算蛋白质组学研究[J].中国科学E辑:技术科学, 2006, 36(2):222-234. |
| SUN R X, FU Y, LI D Q, et al. Computational proteomics based on mass spectrometry [J]. Science in China Series E: Information Sciences, 2006, 36(2): 222-234. | |
| 2 | OLSEN J V, MACEK B, LANGE O, et al. Higher-energy C-trap dissociation for peptide modification analysis [J]. Nature Methods, 2007, 4(9): 709-712. |
| 3 | CHI H, LIU C, YANG H, et al. Comprehensive identification of peptides in tandem mass spectra using an efficient open search engine [J]. Nature Biotechnology, 2018, 36(11): 1059-1061. |
| 4 | CHI H, HE K, YANG B, et al. pFind-Alioth: a novel unrestricted database search algorithm to improve the interpretation of high-resolution MS/MS data[J]. Journal of Proteomics, 2015, 125: 89-97. |
| 5 | WILHELM M, ZOLG D P, GRABER M, et al. Deep learning boosts sensitivity of mass spectrometry-based immunopeptidomics[J]. Nature Communications, 2021, 12: No.3346. |
| 6 | TIWARY S, LEVY R, GUTENBRUNNER P, et al. High-quality MS/MS spectrum prediction for data-dependent and data-independent acquisition data analysis [J]. Nature Methods, 2019, 16(6): 519-525. |
| 7 | VERBRUGGEN S, GESSULAT S, GABRIELS R, et al. Spectral prediction features as a solution for the search space size problem in proteogenomics [J]. Molecular and Cellular Proteomics, 2021, 20: No.100076. |
| 8 | ZHANG Z. Prediction of low-energy collision-induced dissociation spectra of peptides [J]. Analytical Chemistry, 2004, 76(14): 3908-3922. |
| 9 | ZHANG Z. Prediction of low-energy collision-induced dissociation spectra of peptides with three or more charges[J]. Analytical Chemistry, 2005, 77(19): 6364-6373. |
| 10 | SUN S W, YANG F Q, YANG Q, et al. MS-Simulator: predicting y-ion intensities for peptides with two charges based on the intensity ratio of neighboring ions[J]. Journal of Proteome Research, 2012, 11(9): 4509-4516. |
| 11 | WANG Y, YANG F, WU P, et al. OpenMS-Simulator: an open-source software for theoretical tandem mass spectrum prediction [J]. BMC Bioinformatics, 2015, 16: No.110. |
| 12 | ARNOLD R, JAYASANKAR N, AGGARWAL D, et al. A machine learning approach to predicting peptide fragmentation spectra[C]// Proceedings of the 2006 Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing. Singapore: World Scientific Publishing Co Pte Ltd, 2006: 219-230. |
| 13 | LI S, ARNOLD R J, TANG H, et al. On the accuracy and limits of peptide fragmentation spectrum prediction[J]. Analytical Chemistry, 2011, 83(3): 790-796. |
| 14 | DEGROEVE S, MADDELEIN D, MARTENS L. MS2PIP prediction server: compute and visualize MS2 peak intensity predictions for CID and HCD fragmentation [J]. Nucleic Acids Research, 2015, 43(W1): W326-W330. |
| 15 | DEGROEVE S, MARTENS L. MS2PIP: a tool for MS/MS peak intensity prediction[J]. Bioinformatics, 2013, 29(24): 3199-3203. |
| 16 | DONG N P, LIANG Y Z, XU Q S, et al. Prediction of peptide fragment ion mass spectra by data mining techniques[J]. Analytical Chemistry, 2014, 86(15): 7446-7454. |
| 17 | YANG Y, LIN L, QIAO L. Deep learning approaches for data-independent acquisition proteomics[J]. Expert Review of Proteomics, 2021, 18(12): 1031-1043. |
| 18 | WEB B, ZENG W F, LIAO Y, et al. Deep learning in proteomics[J]. Proteomics, 2020, 20(21/22): No.1900335. |
| 19 | MEYER J G. Deep learning neural network tools for proteomics[J]. Cell Reports Methods, 2021, 1(2): No.100003. |
| 20 | ZHOU X X, ZENG W F, CHI H, et al. pDeep: predicting MS/MS spectra of peptides with deep learning[J]. Analytical Chemistry, 2017, 89(23): 12690-12697. |
| 21 | ZENG W F, ZHOU X X, ZHOU W J, et al. MS/MS spectrum prediction for modified peptides using pDeep2 trained by transfer learning [J]. Analytical Chemistry, 2019, 91(15): 9724-9731. |
| 22 | TARN C, ZENG W F. pDeep3: towards more accurate spectrum prediction with fast few-shot learning [J]. Analytical Chemistry, 2021, 93(14): 5815-5822. |
| 23 | ZENG W F, ZHOU X X, WILLEMS S, et al. AlphaPeptDeep: a modular deep learning framework to predict peptide properties for proteomics [J]. Nature Communications, 2022, 13: No.7238. |
| 24 | EKVALL M, TRUONG P, GABRIEL W, et al. Prosit Transformer: a transformer for prediction of MS2 spectrum intensities[J]. Journal of Proteome Research, 2022, 21(5): 1359-1364. |
| 25 | GESSULAT S, SCHMIDT T, ZOLG D P, et al. Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning [J]. Nature Methods, 2019, 16(6): 509-518. |
| 26 | VASWANI A, SHAZEER N M, PARMAR N, et al. Attention is all you need [C]// Proceedings of the 31st International Conference on Neural Information Processing Systems. Red Hook: Curran Associates Inc., 2017:6000-6010. |
| 27 | CHUNG J, GULECEHRE C, CHO K, et al. Empirical evaluation of gated recurrent neural networks on sequence modeling[EB/OL]. [2023-11-11]. . |
| 28 | ZOLG D P, WILHELM M, SCHNATBAUM K, et al. Building proteometools based on a complete synthetic human proteome [J]. Nature Methods, 2017, 14(3): 259-262. |
| 29 | CHICK J M, KOLIPPAKKAM D, NUSINOW D P, et al. A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides [J]. Nature Biotechnology, 2015, 33(7): 743-749. |
| 30 | KULAK N A, PICHLER G, PARON I, et al. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells [J]. Nature Methods, 2014, 11(3): 319-324. |
| 31 | SHARMA K, SCHMITT S, BERGNER C G, et al. Cell type- and brain region-resolved mouse brain proteome[J]. Nature Neuroscience, 2015, 18(12): 1819-1831. |
| 32 | NARAYAN V, LY T, POURKARMI E, et al. Deep proteome analysis identifies age-related processes in C elegans [J]. Cell Systems, 2016, 3(2): 144-159. |
| 33 | YUAN Z F, LIU C, WANG H P, et al. pParse: a method for accurate determination of monoisotopic peaks in high-resolution mass spectra [J]. Proteomics, 2012, 12(2): 226-235. |
| 34 | TYANOVA S, TEMU T, CARLSON A, et al. Visualization of LC-MS/MS proteomics data in MaxQuant [J]. Proteomics, 2015, 15(8): 1453-1456. |
| 35 | LIU K, LI S, WANG L, et al. Full-spectrum prediction of peptides tandem mass spectra using deep neural network[J]. Analytical Chemistry, 2020, 92(6): 4275-4283. |
| 36 | LAPIN J, YAN X, DONG Q. UniSpec: deep learning for predicting the full range of peptide fragment ion series to enhance the proteomics data analysis workflow [J]. Analytical Chemistry, 2024, 96(7): 2783-2790. |
| 37 | COX J. Prediction of peptide mass spectral libraries with machine learning [J]. Nature Biotechnology, 2023, 41(1): 33-43. |
| [1] | Jing QIN, Zhiguang QIN, Fali LI, Yueheng PENG. Diagnosis of major depressive disorder based on probabilistic sparse self-attention neural network [J]. Journal of Computer Applications, 2024, 44(9): 2970-2974. |
| [2] | Xiyuan WANG, Zhancheng ZHANG, Shaokang XU, Baocheng ZHANG, Xiaoqing LUO, Fuyuan HU. Unsupervised cross-domain transfer network for 3D/2D registration in surgical navigation [J]. Journal of Computer Applications, 2024, 44(9): 2911-2918. |
| [3] | Yunchuan HUANG, Yongquan JIANG, Juntao HUANG, Yan YANG. Molecular toxicity prediction based on meta graph isomorphism network [J]. Journal of Computer Applications, 2024, 44(9): 2964-2969. |
| [4] | Yexin PAN, Zhe YANG. Optimization model for small object detection based on multi-level feature bidirectional fusion [J]. Journal of Computer Applications, 2024, 44(9): 2871-2877. |
| [5] | Shunyong LI, Shiyi LI, Rui XU, Xingwang ZHAO. Incomplete multi-view clustering algorithm based on self-attention fusion [J]. Journal of Computer Applications, 2024, 44(9): 2696-2703. |
| [6] | Yuhan LIU, Genlin JI, Hongping ZHANG. Video pedestrian anomaly detection method based on skeleton graph and mixed attention [J]. Journal of Computer Applications, 2024, 44(8): 2551-2557. |
| [7] | Yanjie GU, Yingjun ZHANG, Xiaoqian LIU, Wei ZHOU, Wei SUN. Traffic flow forecasting via spatial-temporal multi-graph fusion [J]. Journal of Computer Applications, 2024, 44(8): 2618-2625. |
| [8] | Qianhong SHI, Yan YANG, Yongquan JIANG, Xiaocao OUYANG, Wubo FAN, Qiang CHEN, Tao JIANG, Yuan LI. Multi-granularity abrupt change fitting network for air quality prediction [J]. Journal of Computer Applications, 2024, 44(8): 2643-2650. |
| [9] | Zheng WU, Zhiyou CHENG, Zhentian WANG, Chuanjian WANG, Sheng WANG, Hui XU. Deep learning-based classification of head movement amplitude during patient anaesthesia resuscitation [J]. Journal of Computer Applications, 2024, 44(7): 2258-2263. |
| [10] | Huanhuan LI, Tianqiang HUANG, Xuemei DING, Haifeng LUO, Liqing HUANG. Public traffic demand prediction based on multi-scale spatial-temporal graph convolutional network [J]. Journal of Computer Applications, 2024, 44(7): 2065-2072. |
| [11] | Zhi ZHANG, Xin LI, Naifu YE, Kaixi HU. DKP: defending against model stealing attacks based on dark knowledge protection [J]. Journal of Computer Applications, 2024, 44(7): 2080-2086. |
| [12] | Yiqun ZHAO, Zhiyu ZHANG, Xue DONG. Anisotropic travel time computation method based on dense residual connection physical information neural networks [J]. Journal of Computer Applications, 2024, 44(7): 2310-2318. |
| [13] | Song XU, Wenbo ZHANG, Yifan WANG. Lightweight video salient object detection network based on spatiotemporal information [J]. Journal of Computer Applications, 2024, 44(7): 2192-2199. |
| [14] | Xun SUN, Ruifeng FENG, Yanru CHEN. Monocular 3D object detection method integrating depth and instance segmentation [J]. Journal of Computer Applications, 2024, 44(7): 2208-2215. |
| [15] | Yajuan ZHAO, Fanjun MENG, Xingjian XU. Review of online education learner knowledge tracing [J]. Journal of Computer Applications, 2024, 44(6): 1683-1698. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||