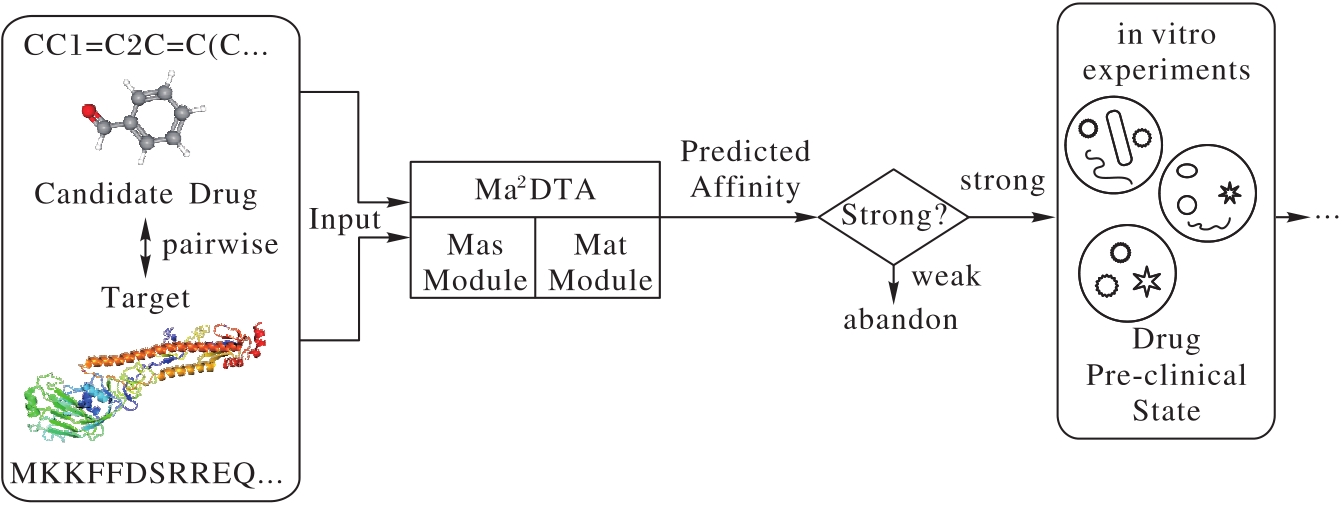
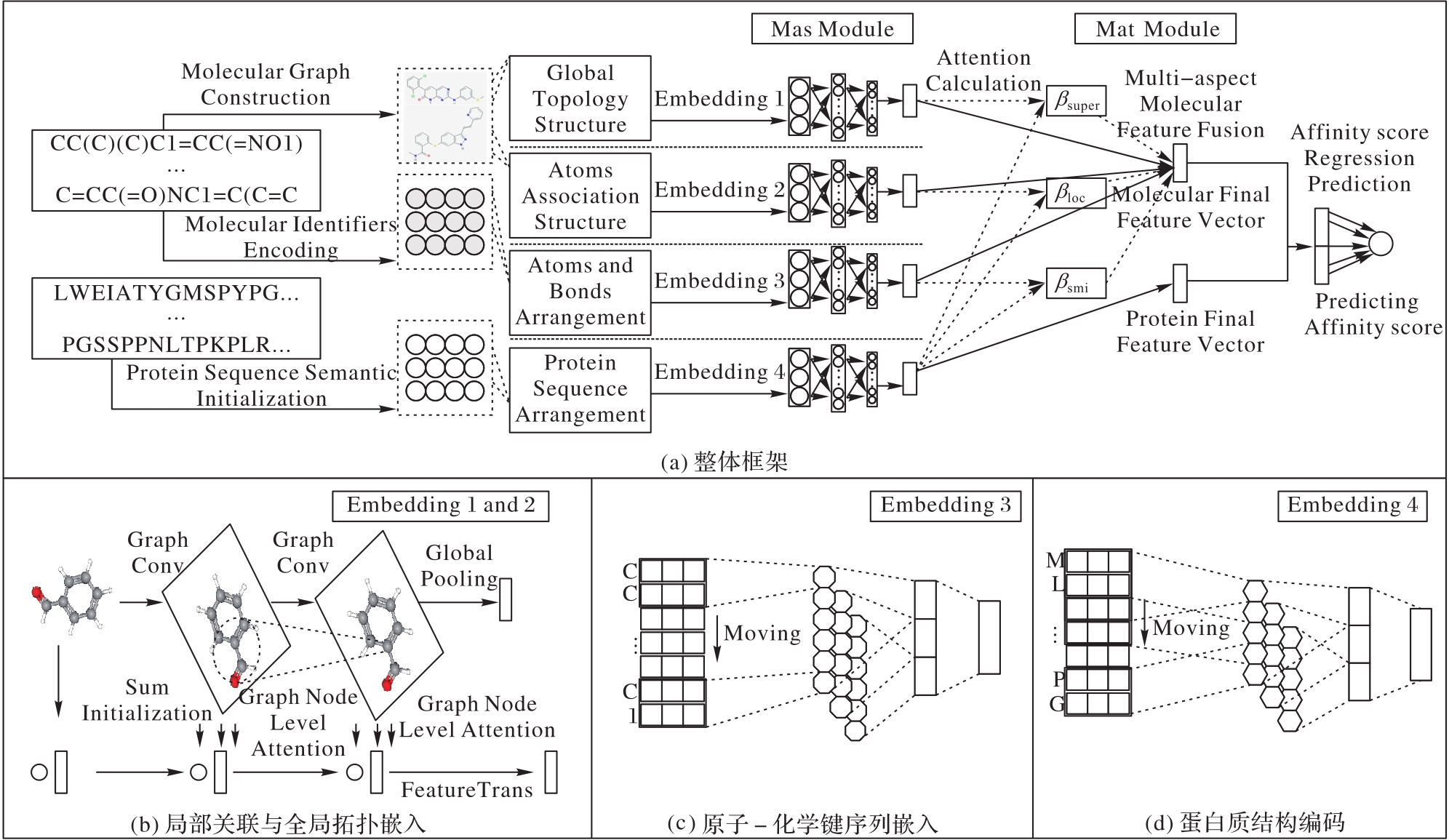
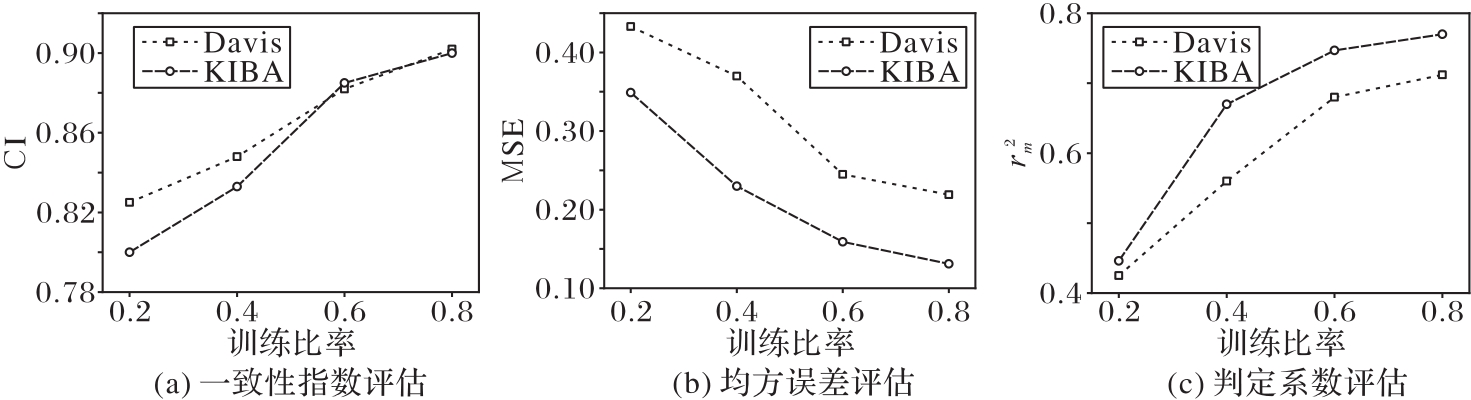
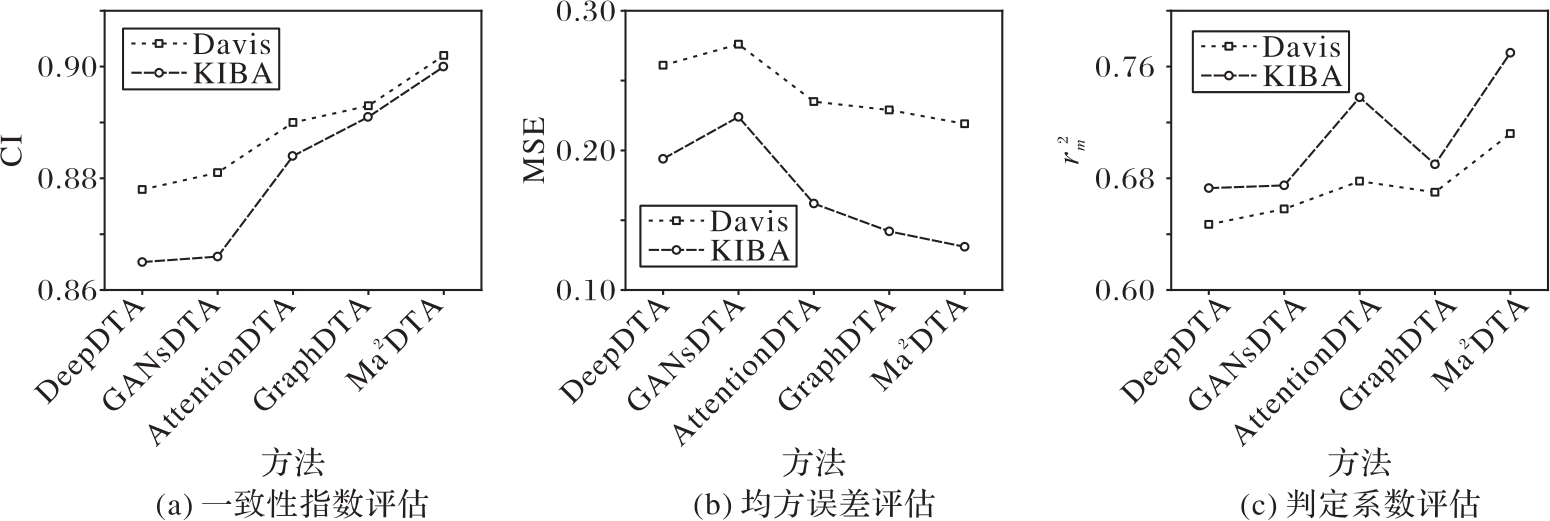
| 1 |
BAGHERIAN M, SABETI E, WANG K, et al. Machine learning approaches and databases for prediction of drug-target interaction: a survey paper[J]. Briefings in Bioinformatics, 2021, 22(1): 247-269. 10.1093/bib/bbz157
|
| 2 |
LU S, YE Q Z, SINGH D, et al. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein[J]. Nature Communications, 2021, 12: No.502. 10.1101/2020.07.30.228023
|
| 3 |
POLAND G A. Tortoises, hares, and vaccines: a cautionary note for SARS-CoV-2 vaccine development[J]. Vaccine, 2020, 38(27): 4219-4220. 10.1016/j.vaccine.2020.04.073
|
| 4 |
BOLTON E E, WANG Y L, THIESSEN P A, et al. PubChem: integrated platform of small molecules and biological activities[M]// WHEELER R A, SPELLMEYER D C. Annual Reports in Computational Chemistry, Volume 4. New York: Elsevier Science, 2008: 217-241.
|
| 5 |
NOBLE M E M, ENDICOTT J A, JOHNSON L N. Protein kinase inhibitors: insights into drug design from structure[J]. Science, 2004, 303(5665): 1800-1805. 10.1126/science.1095920
|
| 6 |
LIN X. DeepGS: deep representation learning of graphs and sequences for drug-target binding affinity prediction[EB/OL]. (2020-04-03) [2021-02-20].. 10.1093/bib/bbab117
|
| 7 |
徐冰冰,岑科廷,黄俊杰,等. 图卷积神经网络综述[J]. 计算机学报, 2020, 43(5):755-780. 10.11897/SP.J.1016.2020.00755
|
|
XU B B, CEN K T, HUANG J J, et al. A survey on graph convolutional neural network[J]. Chinese Journal of Computers, 2020, 43(5):755-780. 10.11897/SP.J.1016.2020.00755
|
| 8 |
TROTT O, OLSON A J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading[J]. Journal of Computational Chemistry, 2010, 31(2): 455-461.
|
| 9 |
PERLMAN L, GOTTLIEB A, ATIAS N, et al. Combining drug and gene similarity measures for drug-target elucidation[J]. Journal of Computational Biology, 2011, 18(2): 133-145. 10.1089/cmb.2010.0213
|
| 10 |
WANG M H, TANG C, CHEN J J. Drug-target interaction prediction via dual Laplacian graph regularized matrix completion[J]. BioMed Research International, 2018, 2018: No.1425608. 10.1155/2018/1425608
|
| 11 |
HE T, HEIDEMEYER M, BAN F Q, et al. SimBoost: a read-across approach for predicting drug-target binding affinities using gradient boosting machines[J]. Journal of Cheminformatics, 2017, 9: No.24. 10.1186/s13321-017-0209-z
|
| 12 |
GAWEHN E, HISS J A, SCHNEIDER G. Deep learning in drug discovery[J]. Molecular Informatics, 2016, 35(1): 3-14. 10.1002/minf.201501008
|
| 13 |
SCHÜTT K T, ARBABZADAH F, CHMIELA S, et al. Quantum-chemical insights from deep tensor neural networks[J]. Nature Communications, 2017, 8: No.13890. 10.1038/ncomms13890
|
| 14 |
CALLAWAY E. ‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures[J]. Nature, 2020, 588(7837): 203-204. 10.1038/d41586-020-03348-4
|
| 15 |
WEN M, ZHANG Z M, NIU S Y, et al. Deep-learning-based drug-target interaction prediction[J]. Journal of Proteome Research, 2017, 16(4): 1401-1409. 10.1021/acs.jproteome.6b00618
|
| 16 |
ÖZTÜRK H, ÖZGÜR A, OZKIRIMLI E. DeepDTA: deep drug-target binding affinity prediction[J]. Bioinformatics, 2018, 34(17): i821-i829. 10.1093/bioinformatics/bty593
|
| 17 |
KARIMI M, WU D, WANG Z Y, et al. DeepAffinity: interpretable deep learning of compound-protein affinity through unified recurrent and convolutional neural networks[J]. Bioinformatics, 2019, 35(18): 3329-3338. 10.1093/bioinformatics/btz111
|
| 18 |
JIMÉNEZ-LUNA J, GRISONI F, SCHNEIDER G. Drug discovery with explainable artificial intelligence[J]. Nature Machine Intelligence, 2020, 2(10): 573-584. 10.1038/s42256-020-00236-4
|
| 19 |
GAO K Y K, FOKOUE A, LUO H, et al. Interpretable drug target prediction using deep neural representation[C]// Proceedings of the 27th International Joint Conference on Artificial Intelligence. [S.l.]: ijcai.org, 2018: 3371-3377. 10.24963/ijcai.2018/468
|
| 20 |
NGUYEN T, LE H, QUINN T P, et al. GraphDTA: predicting drug-target binding affinity with graph neural networks[J]. Bioinformatics, 2021, 37(8): 1140-1147. 10.1093/bioinformatics/btaa921
|
| 21 |
LI S Y, WAN F P, SHU H T, et al. MONN: a multi-objective neural network for predicting compound-protein interactions and affinities[J]. Cell Systems, 2020, 10(4): 308-322.e11. 10.1016/j.cels.2020.03.002
|
| 22 |
ISHIGURO K, MAEDA S I, KOYAMA M. Graph warp module: an auxiliary module for boosting the power of graph neural networks in molecular graph analysis[EB/OL]. (2019-05-24) [2021-01-03]..
|
| 23 |
KIPF T N, WELLING M. Semi-supervised classification with graph convolutional networks[EB/OL]. (2017-02-22) [2020-03-20]..
|
| 24 |
DAVIS M I, HUNT J P, HERRGARD S, et al. Comprehensive analysis of kinase inhibitor selectivity[J]. Nature Biotechnology, 2011, 29(11): 1046-1051. 10.1038/nbt.1990
|
| 25 |
TANG J, SZWAJDA A, SHAKYAWAR S, et al. Making sense of large-scale kinase inhibitor bioactivity data sets: a comparative and integrative analysis[J]. Journal of Chemical Information and Modeling, 2014, 54(3): 735-743. 10.1021/ci400709d
|
| 26 |
ROY K, CHAKRABORTY P, MITRA I, et al. Some case studies on application of “rm2” metrics for judging quality of quantitative structure-activity relationship predictions: emphasis on scaling of response data[J]. Journal of Computational Chemistry, 2013, 34(12): 1071-1082. 10.1002/jcc.23231
|
| 27 |
ZHAO Q C, XIAO F, YANG M Y, et al. AttentionDTA: prediction of drug-target binding affinity using attention model[C]// Proceedings of 2019 IEEE International Conference on Bioinformatics and Biomedicine. Piscataway: IEEE, 2019: 64-69. 10.1109/bibm47256.2019.8983125
|
| 28 |
ZHAO L L, WANG J J, PANG L, et al. GANsDTA: predicting drug-target binding affinity using GANs[J]. Frontiers in Genetics, 2020, 10: No.1243. 10.3389/fgene.2019.01243
|


 ), 秦琪琦1, 张泽华1, 郭旭敏2
), 秦琪琦1, 张泽华1, 郭旭敏2